









Emma B. Hodcroft

Much of the analysis of these genome sequences is not done by public-health bodies. It rests on the initiative of academic researchers, many of them early in their careers, who cobble together software and analytical tools in their own time to find essential answers. Nextstrain1, an open-source project involving groups from Switzerland and the United States, is helping to coordinate these efforts. One of us (E.B.H.), a Nextstrain researcher, has been working to track variants since September 2020 (see https://nextstrain.org/ncov/global). Less than two hours after the spread of an alarming new variant (now called 501Y.V1, or B.1.1.7) was announced by the UK health minister in December 2020, E.B.H. had provided context for its key mutations in a series of tweets, and showed its progression in the United Kingdom and across Europe in the months before (see go.nature.com/3ptrya5). The Twitter thread became a key source of information on the new variant, and E.B.H.’s Christmas break was lost to crunching further sequences and briefing journalists.
The ‘phylogenetic’ tools used to track these variants were largely developed by evolutionary biologists to study the lineage of organisms. They were designed to construct phylogenetic trees that can ask, for example, whether flight evolved twice in mammals, or whether two large groups of bats began as one that then diverged.
Now, co-opted to track a pandemic, the leaves of the phylogenetic tree represent SARS-CoV-2 sequences taken from individuals, and the tree grows with the number of cases (see ‘Mutation watch’). With more than 5,000 sequences coming in each day (and rising), the tools can quickly help to determine whether an observed mutation has changed the biology of the virus. Such information is crucial to designers of drugs, vaccines and policies.
However, as these data continue to flood in, keeping the phylogenetic trees up to date is becoming increasingly difficult. Nextstrain was previously used to track influenza and Ebola outbreaks, but often retrospectively or through small updates every week or month — not to track thousands of sequences a day during the peak of a global pandemic. Now, researchers need to update their analyses daily.
Here we describe how phylogenetics can help in understanding SARS-CoV-2 outbreaks and variants. We outline the technical and institutional challenges of automating this painstaking detective work. As experts in bioinformatics and viral evolution, we know that our tools are buckling under the pressure when they are most needed. We must move beyond the limitations of existing tools and improve processes, so that they are fit to handle a pandemic.
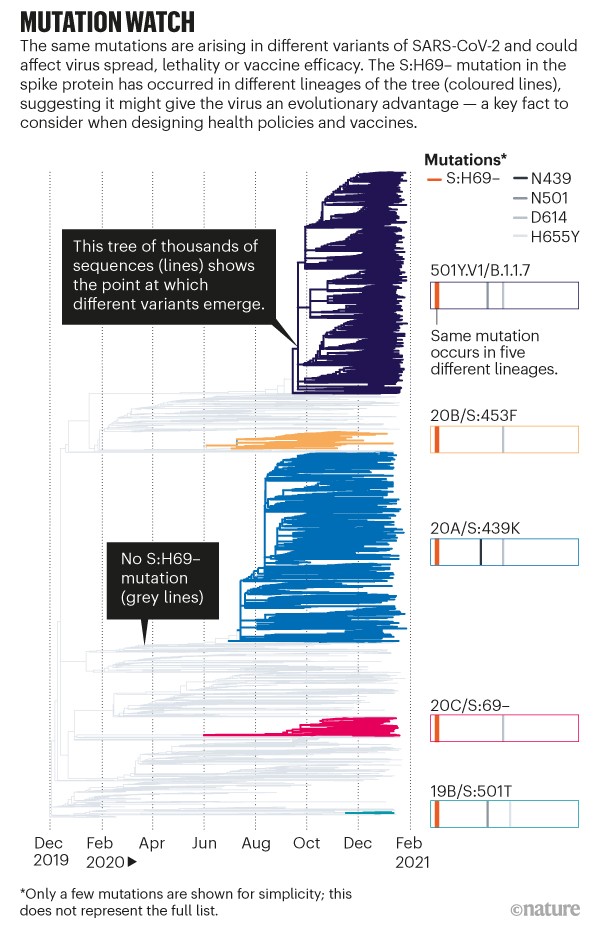
Source: GISAID
Three uses
What can public-health officials do with viral phylogenetics?
Track transmission. To stall a pandemic, researchers need to track who caught what from whom. This enables them to answer such questions as: what makes a superspreading event? Which public-health practices slow transmission? Are precautions working?
On 29 September last year, a passenger in Dubai who had travelled from Switzerland, and was in the early, infectious stage of COVID-19 despite testing negative, boarded a plane to New Zealand and seems to have infected at least four fellow passengers2. Working that out required too many people — at least 26 researchers — and too much luck. Had the infective viral strain come from a cluster in the United States, say, rather than from Switzerland, researchers probably wouldn’t have had the correct data to be able to tell. This reflects the patchy sequencing in different countries.
Furthermore, it is hard to reconstruct transmission histories from samples alone because many of the sequences are identical. To succeed in the Dubai case (a rare example of a peer-reviewed scientific paper combining phylogenies with an aircraft seating plan), the investigators integrated genomic analysis with knowledge about who was diagnosed when and where, and who they were in contact with. Phylogenetics was key to public understanding that air travel was riskier than many understood.
Tracking how two superspreader events in Boston, Massachusetts, varied in their geographical spread, and determining the amount of mutations generated, required the work of more than 50 researchers. Again, this was possible only because of unusual circumstances: dense, early sampling in that area3.
Flag key mutations. Knowing which variants to pay attention to could be the difference between containing an outbreak or speeding up its spread. A country might wish to change its vaccination strategy if dominant variants are thought to reduce vaccine efficacy, as has been the case in South Africa. Phylogenetics, especially combined with in vitro work, can probe whether a mutation is likely to have changed the biology of the virus. Subtler signals are worth finding, too. Individuals infected with certain strains might one day receive specific medical treatment or be managed differently in quarantine.
In late 2020, 17 million mink were marked for culling when a new combination of mutations arose in a variant in Danish mink farms that showed reduced antibody binding. The variant never spread beyond 11 people. If it had been easier to link up the observed mutations with their expected impact on the virus and their apparent link to adaptation in mink (rather than humans), could this have been navigated more smoothly?
So far, phylogenetic analyses tend to operate separately from wet labs. For example, Datamonkey is a collection of modelling and bioinformatics tools run by researchers at Temple University in Philadelphia, Pennsylvania. They developed a web tool that scans SARS-CoV-2 phylogenies daily to identify signatures of natural selection (http://covid19.datamonkey.org). Likewise, computational biologist Lucy van Dorp and her colleagues at University College London mine genomic databases for mutations associated with increased viral spread4. However, computational analyses might not always be tested experimentally. Meanwhile, lab-based experiments investigating these different variants and their responses to vaccines in cell cultures5 are rarely combined with phylogenies.
We need ways to combine data from wet biology and sequence analysis to produce a joined-up picture of mutations as they emerge and spread.
Estimate R. Phylogenetic information can improve estimates of this key metric of the pandemic: the average number of new infections each infected person causes in a population that has some immunity, called the effective reproduction number (Re). How? Phylogeny can distinguish between viruses that have been imported into a community and those that have spread within it. Australia, parts of the United States and New Zealand have incorporated such measurements into Re and related metrics. Methods that combined information on epidemiology with phylogenetic data were used to show that a lockdown in New Zealand reduced Re from 7 at the start of one outbreak to 0.2 at the end of March 2020 in one cluster of cases6.
Despite their power, these methods are rarely used. They are complex to produce and require expertise that is in short supply. Training and more user-friendly software would help to expand phylogenetic epidemiology.
Upgrades needed
Accommodate and communicate uncertainty. Some parts of the evolutionary history of SARS-CoV-2 are more certain than others. Researchers are confident in the deeper branching near the root of the tree, showing the virus’s spread from China in early 2020. It is much less clear whether near-identical sequences represent a direct transmission.
Yet phylogenies could be used to say with some certainty who did not infect a particular individual, and so assess transmission risk. Unfortunately, this confidence in non-relatedness is not captured by any conventional measures, making it hard for public-health officials to use phylogenies to make quick decisions. Recent work proposes a new way to quantify uncertainty in response to this problem7, but there is much more to be done.
To trace the path of the virus, we can use sophisticated methods that combine phylogenetic uncertainty, models of transmission, and patient and sequence data. However, these methods are currently much too computationally intensive to be used for each sample gathered. In the initial epidemic wave, computational biologist Louis du Plessis at the University of Oxford, UK, and his colleagues used thousands of SARS-CoV-2 sequences to find more than 1,000 genetically distinct chains of infection within the United Kingdom8. But to do so, they had to come up with specific shortcuts and simplifying assumptions that might not work in other contexts.

Workers distribute COVID-19 test kits to residents of Maidstone, UK, in a bid to track a fast-spreading variant first identified in South Africa.Credit: Chris Ratcliffe/Bloomberg via Getty
Find dodgy data. The rush to share data, and the (necessary) entry of less-experienced labs into sequencing have meant that data can be full of small but dangerous errors — in the sequences themselves and in the location and timing ‘metadata’ that go with them. At the start of this year, scientists noticed an increase in sequences labelled January 2020, around the time the pandemic began. Were new samples being dug out of freezers in search of insightful clues? Unfortunately, the culprit was a hard-coded ‘2020’ in the processing scripts that hadn’t been updated as 2021 began.
In the sequences themselves, errors can be even more subtle, and more impactful. Contamination, poor-quality samples and mistakes that creep in during the processing pipeline can introduce false mutations or even remove real ones. These errors then propagate into downstream analyses, redrawing connections that can mislead outbreak investigations or get blamed for changes to the biology of the virus. There are individual efforts to root out the most troublesome parts of the data9. But what scientists really need are stable and open infrastructures that allow the entire community to correct sequences and metadata throughout the pandemic.
Accommodate uneven sampling. The United Kingdom has so far sequenced almost 5,000 samples per 100,000 cases, compared with 320 per 100,000 in the United States and 30 per 100,000 in Brazil (see go.nature.com/3qutqwj). Even in the United Kingdom, certain regions are better represented in sequencing data than are others. The tools we have to infer geographical spread from samples do not typically account for these biases. Without correction, the United Kingdom would be labelled a common source of variants just because of its high rate of sequencing, not because of how variants are spreading. Researchers can account for this in rough ways, but not on a detailed level (see ‘Uneven sequencing’).
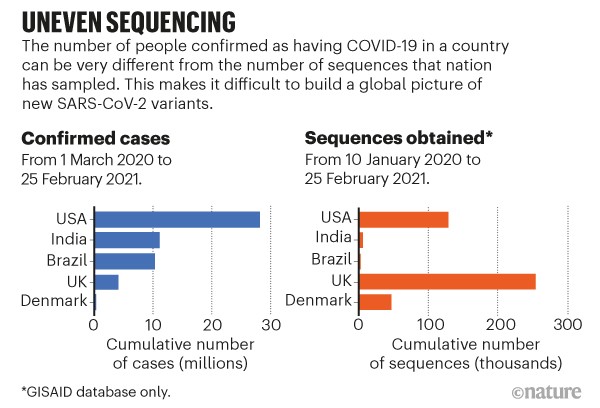
Sources: Our World in Data (cases)/GISAID (sequences)
The start of this year has led to yet another type of sampling bias that must be accounted for. One of the three main variants of concern, 501Y.V1 (or B.1.1.7), has a deletion in the spike protein. This causes the diagnostic PCR test to fail to detect the spike gene — a problem called S drop-out. But 501Y.V1 is not the only variant that causes such drop-outs, so confirmation can come only from sequencing. Some countries are now preferentially sequencing S drop-outs in an effort to spot and track a more-transmissible variant. In the process, they’re biasing their data towards particular variants, meaning such data might no longer represent the true proportions present.
The methods proposed to account for these biases take too long to run with large data sets. Worse, many analyses assume that the population of the virus is stable — it is not. Ideally, computational tools would be robust to sampling bias, and would combine with databases that allow scientists to record why each sample was sequenced. This could help to identify the growth and geographical origin of new variants, and help public-health officials to overcome sampling issues.
Community fixes
Frictionless data. Much progress has been made in data sharing, but we must continue the conversation10,11 (see also go.nature.com/37jgthu).
Databases provide crucial curatorial services. Submission speed, ease of contributing data and incredible collation efforts have made several repositories invaluable to scientists and public-health practitioners around the world. To encourage timely sharing, provisions to ensure data submitters are credited and rewarded for their work and protected from having their analyses scooped are crucial.
However, the pandemic has provided an opportunity to examine how data access and ecosystems for open data analysis could be harnessed for even more benefit. Protecting data submitters, for example, should be carefully considered against the potential to hinder data reuse and critical analyses in ways that might not have been anticipated12.
Stakeholders should craft recommendations — for data interoperability, management and scalability, among other things. As a community, we need to agree on how to balance openness and credit to get the best for public health.
Rethink incentives
Scientists’ reward systems almost punish them for public service during the pandemic. Academics have been informally seconded to assist with public-health efforts and to participate in government task forces. They are neglecting their own research, creating ‘goodwill gaps’ in their publication records.
Because of the relentless urgency of the crisis, the scientists who are doing the most work for public health have the least time to apply for the grants and jobs that sustain them. This predicament disproportionately affects early-career researchers, who are typically on short contracts. Institutions should amend their assessment criteria to recognize this labour and intellectual flexibility.
Researchers and public-health officials need to work together to make tools easier to use, and to train others. Secondments should be funded by governments, making it easier for researchers to step away from their academic work during public-health emergencies. (After this pandemic, perhaps some of these temporary positions should become permanent.)
What now?
With enough support to develop and deploy the right tools, phylogenetics researchers can detect emerging SARS-CoV-2 variants and rapidly reconstruct an outbreak’s transmission history. We call on researchers, funders and public-health institutions, nationally and internationally, to put in place the resources, incentives and mandates to build phylogenetic and public-health collaborations for the benefit of all.
No comments:
Post a Comment